Platelet
| Platelets | |
|---|---|
 Image from a light microscope (500 ×) from a Giemsa-stained peripheral blood smear showing platelets (blue dots) surrounded by red blood cells (pink circular structures) | |
| Details | |
| Precursor | Megakaryocytes |
| Function | Formation of blood clots; prevention of bleeding |
| Identifiers | |
| Latin | Thrombocytes |
Anatomical terms of microanatomy [edit on Wikidata] | |
Platelets, also called thrombocytes (from Greek θρόμβος, "clot" and κύτος, "cell"), are a component of blood whose function (along with the coagulation factors) is to react to bleeding from blood vessel injury by clumping, thereby initiating a blood clot.[1] Platelets have no cell nucleus: they are fragments of cytoplasm that are derived from the megakaryocytes[2] of the bone marrow, and then enter the circulation. Circulating unactivated platelets are biconvex discoid (lens-shaped) structures,[3][4]:117–18 2–3 µm in greatest diameter.[5] Platelets are found only in mammals, whereas in other animals (e.g. birds, amphibians) thrombocytes circulate as intact mononuclear cells.[4]:3
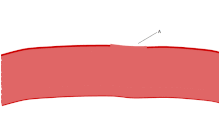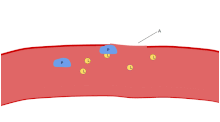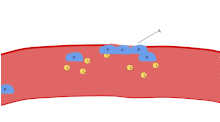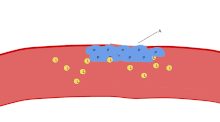
The ligands, denoted by letter L, signal for platelets (P) to migrate towards the wound (Site A). As more platelets gather around the opening, they produce more ligands to amplify the response. The platelets congregate around the wound in order to create a cap to stop blood flow out of the tissue.
On a stained blood smear, platelets appear as dark purple spots, about 20% the diameter of red blood cells. The smear is used to examine platelets for size, shape, qualitative number, and clumping. The ratio of platelets to red blood cells in a healthy adult ranges from 1:10 to 1:20.
One major function of platelets is to contribute to hemostasis: the process of stopping bleeding at the site of interrupted endothelium. They gather at the site and unless the interruption is physically too large, they plug the hole. First, platelets attach to substances outside the interrupted endothelium: adhesion. Second, they change shape, turn on receptors and secrete chemical messengers: activation. Third, they connect to each other through receptor bridges: aggregation.[6] Formation of this platelet plug (primary hemostasis) is associated with activation of the coagulation cascade with resultant fibrin deposition and linking (secondary hemostasis). These processes may overlap: the spectrum is from a predominantly platelet plug, or "white clot" to a predominantly fibrin, or "red clot" or the more typical mixture. Some would add the subsequent retraction and platelet inhibition as fourth and fifth steps to the completion of the process[7] and still others a sixth step wound repair. Platelets also participate in both innate[8] and adaptive[9] intravascular immune responses.
Low platelet concentration is called thrombocytopenia, and is due to either decreased production or increased destruction. Elevated platelet concentration is called thrombocytosis, and is either congenital, reactive (to cytokines), or due to unregulated production: one of the myeloproliferative neoplasms or certain other myeloid neoplasms. A disorder of platelet function is a thrombocytopathy.
Normal platelets can respond to an abnormality on the vessel wall rather than to hemorrhage, resulting in inappropriate platelet adhesion/activation and thrombosis: the formation of a clot within an intact vessel. This type of thrombosis arises by mechanisms different than those of a normal clot: namely, extending the fibrin of venous thrombosis; extending an unstable or ruptured arterial plaque, causing arterial thrombosis; and microcirculatory thrombosis. An arterial thrombus may partially obstruct blood flow, causing downstream ischemia, or may completely obstruct it, causing downstream tissue death.
Contents
1 Measurement
2 Structure
3 Development
4 Hemostasis
4.1 Adhesion
4.2 Activation
4.2.1 Inhibition
4.2.2 Trigger (induction)
4.2.3 Components (consequences)
4.2.3.1 GPIIb/IIIa activation
4.2.3.2 Granule secretion
4.2.3.3 Morphology change
4.2.3.4 Platelet-coagulation factor interactions: coagulation facilitation
4.3 Aggregation
4.4 Wound repair
5 Immune function
5.1 Immunothrombosis
5.2 Inflammation
5.3 Adaptive immunity
6 Signs and symptoms of disorders
7 Tests of function
7.1 Bleeding time
7.2 Multiplate multiple electrode aggregometry
7.3 PFA-100
8 Disorders
8.1 Thrombocytopenia
8.2 Altered platelet function
8.3 Thrombocytosis and thrombocythemia
9 Drugs affecting
9.1 Anti-inflammatory drugs
9.2 Drugs that suppress platelet function
9.2.1 Oral agents
9.3 Drugs that stimulate platelet production
9.3.1 Intravenous agents
10 Therapy with platelets
10.1 Transfusion
10.1.1 Indications
10.1.2 Collection
10.1.3 Storage
10.1.4 Delivery to recipients
10.2 Wound therapy
11 Other animals
12 History
13 References
14 External links
Measurement
Platelet concentration is measured either manually using a hemocytometer, or by placing blood in an automated platelet analyzer using electrical impedance, such as a Coulter counter.[10] The normal range (99% of population analyzed) for platelets in healthy Caucasians is 150,000 to 450,000 per cubic millimeter [11](a mm3 equals a microliter). or 150–450 × 109 per liter. The normal range has been confirmed to be the same in the elderly[12] and Spanish populations.[13]
Structure
Structurally the platelet can be divided into four zones, from peripheral to innermost:
- Peripheral zone – is rich in glycoproteins required for platelet adhesion, activation, and aggregation. For example, GPIb/IX/X; GPVI; GPIIb/IIIa.
- Sol-gel zone – is rich in microtubules and microfilaments, allowing the platelets to maintain their discoid shape.
- Organelle zone – is rich in platelet granules. Alpha granules contain clotting mediators such as factor V, factor VIII, fibrinogen, fibronectin, platelet-derived growth factor, and chemotactic agents. Delta granules, or dense bodies, contain ADP, calcium, serotonin, which are platelet-activating mediators.
- Membranous zone – contains membranes derived from megakaryocytic smooth endoplasmic reticulum organized into a dense tubular system which is responsible for thromboxane A2 synthesis. This dense tubular system is connected to the surface platelet membrane to aid thromboxane A2 release.
Development

Platelets derive from totipotent marrow stem cells
Megakaryocyte and platelet production is regulated by thrombopoietin, a hormone produced in the kidneys and liver.- Each megakaryocyte produces between 1,000 and 3,000 platelets during its lifetime.
- An average of 1011 platelets are produced daily in a healthy adult.
- Reserve platelets are stored in the spleen, and are released when needed by splenic contraction induced by the sympathetic nervous system.

Platelets extruded from megakaryocytes
- The average life span of circulating platelets is 8 to 9 days.[14] Life span of individual platelets is controlled by the internal apoptotic regulating pathway, which has a Bcl-xL timer.[15]
- Old platelets are destroyed by phagocytosis in the spleen and liver.
Hemostasis

3D rendering of four inactivated and three activated platelets.
An overview summarizing platelet dynamics, the complex process of converting inactive platelets into a platelet plug, is essentialEL 2. Complicating any verbal description is the fact that at least 193 proteins and 301 interactions are involved in platelet dynamics. The separation of platelet dynamics into three stages is useful in this regard, but it is artificial: in fact, each stage is initiated in rapid succession, and each continues until the trigger for that stage is no longer present, so there is overlap.[6]
Adhesion
Thrombus formation on an intact endothelium is prevented by nitric oxide,[16]prostacyclin,[17] and CD39.[18]
Endothelial cells are attached to the subendothelial collagen by von Willebrand factor (VWF) which these cells produce. VWF is also stored in the Weibel-Palade bodies of the endothelial cells and secreted constitutively into the blood. Platelets store vWF in their alpha granules.
When the endothelial layer is disrupted, collagen and VWF anchor platelets to the subendothelium. Platelet GP1b-IX-V receptor binds with VWF; and GPVI receptor and integrin α2β1 bind with collagen.[19]
Activation

Scanning electron micrograph of blood cells. From left to right: human erythrocyte, activated platelet, leukocyte.
Inhibition
The intact endothelial lining inhibits platelet activation by producing nitric oxide, endothelial-ADPase, and PGI2 (Prostacyclin). Endothelial-ADPase degrades the platelet activator ADP.
Resting platelets maintain active calcium efflux via a cyclic AMP activated calcium pump. Intracellular calcium concentration determines platelet activation status, as it is the second messenger that drives platelet conformational change and degranulation (see below). Endothelial prostacyclin binds to prostanoid receptors on the surface of resting platelets. This event stimulates the coupled Gs protein to increase adenylate cyclase activity and increases the production of cAMP, further promoting the efflux of calcium and reducing intracellular calcium availability for platelet activation.
ADP on the other hand binds to purinergic receptors on platelet surface. Since the thrombocytic purinergic receptor P2Y12 is coupled to Gi proteins, ADP reduces platelet adenylate cyclase activity and cAMP production, leading to accumulation of calcium inside the platelet by inactivating the cAMP calcium efflux pump. The other ADP-receptor P2Y1 couples to Gq that activates phospholipase C-beta 2 PLCB2, resulting in inositol 1,4,5-trisphosphate (IP3)generation and intracellular release of more calcium. This together induces platelet activation. Endothelial ADPase degrades ADP and prevents this from happening. Clopidogrel and related antiplatelet medications also work as purinergic receptor P2Y12 antagonists.
Trigger (induction)
Platelet activation begins seconds after adhesion occurs. It is triggered when collagen from the subendothelium binds with its receptors (GPVI receptor and integrin α2β1) on the platelet. GPVI is associated with the Fc receptor gamma chain and leads via the activation of a tyrosine kinase cascade finally to the activation of PLC-gamma2 PLCG2 and more calcium release.
Tissue factor also binds to factor VII in the blood, which initiates the intrinsic coagulation cascade to increase thrombin production. Thrombin is a potent platelet activator, acting through Gq and G12. These are G protein coupled receptors and they turn on calcium mediated signaling pathways within the platelet, overcoming the baseline calcium efflux. Families of three G proteins (Gq, Gi, G12) operate together for full activation. Thrombin also promotes secondary fibrin-reinforcement of the platelet plug. Platelet activation in turn degranulates and releases factor V and fibrinogen, potentiating the coagulation cascade. So in reality the process of platelet plugging and coagulation are occurring simultaneously rather than sequentially, with each inducing the other to form the final .
Components (consequences)
GPIIb/IIIa activation
Collagen-mediated GPVI signalling increases the platelet production of thromboxane A2 (TXA2) and decreases the production of prostacyclin. This occurs by altering the metabolic flux of platelet's eicosanoid synthesis pathway, which involves enzymes phospholipase A2, cyclo-oxygenase 1, and thromboxane-A synthase. Platelets secrete thromboxane A2, which acts on the platelet's own thromboxane receptors on the platelet surface (hence the so-called "out-in" mechanism), and those of other platelets. These receptors trigger intraplatelet signaling, which converts GPIIb/IIIa receptors to their active form to initiate aggregation.[6]
Granule secretion

Diagram of the structure of a platelet showing the granules
Platelets contain dense granules, lambda granules and alpha granules. Activated platelets secrete the contents of these granules through their canalicular systems to the exterior. Simplistically, bound and activated platelets degranulate to release platelet chemotactic agents to attract more platelets to the site of endothelial injury. Granule characteristics:
α granules (alpha granules) – containing P-selectin, platelet factor 4, transforming growth factor-β1, platelet-derived growth factor, fibronectin, B-thromboglobulin, vWF, fibrinogen, and coagulation factors V and XIII.
δ granules (delta or dense granules) – containing ADP or ATP, calcium, and serotonin.- γ granules (gamma granules) – similar to lysosomes and contain several hydrolytic enzymes.
- λ granules (lambda granules) – contents involved in resorption during later stages of vessel repair.
Morphology change
Mitochondrial hyperpolarization is a key event in initiating changes in morphology.[20] Intraplatelet calcium concentration increases, stimulating the interplay between the microtubule/actin filament complex. The continuous changes in shape from the unactivated to the fully activated platelet is best seen on scanning electron microscopy. Three steps along this path are named early dendritic, early spread and spread. The surface of the unactivated platelet looks very similar to the surface of the brain, with a wrinkled appearance from numerous shallow folds to increase the surface area; early dendritic, an octopus with multiple arms and legs; early spread, an uncooked frying egg in a pan, the "yolk" being the central body; and the spread, a cooked fried egg with a denser central body.
These changes are all brought about by the interaction of the microtubule/actin complex with the platelet cell membrane and open canalicular system (OCS), which is an extension and invagination of that membrane. This complex runs just beneath these membranes, and is the chemical motor which literally pulls the invaginated OCS out of the interior of the platelet, like turning pants pockets inside out, creating the dendrites. This process is similar to the mechanism of contraction in a muscle cell.[21]. The entire OCS thus becomes indistinguishable from the initial platelet membrane as it forms the "fried egg". This dramatic increase in surface area comes about with neither stretching nor adding phospholipids to the platelet membrane.[22]
Platelet-coagulation factor interactions: coagulation facilitation
Platelet activation causes its membrane surface to become negatively charged. One of the signaling pathways turns on scramblase, which moves negatively charged phospholipids from the inner to the outer platelet membrane surface. These phospholipids then bind the tenase and prothrombinase complexes, two of the sites of interplay between platelets and the coagulation cascade. Calcium ions are essential for the binding of these coagulation factors.
In addition to interacting with vWF and fibrin, platelets interact with thrombin, Factors X, Va, VIIa, XI, IX, and prothrombin to complete formation via the coagulation cascade.[23][24]
Six studies suggested platelets express tissue factor: the definitive study shows they do not.[23] The platelets from rats were conclusively shown to express tissue factor protein and also it was proved that the rat platelets carry both the tissue factor pre-mRNA and mature mRNA.[25]
Aggregation

Platelet clumps in a blood smear
Aggregation begins minutes after activation, and occurs as a result of turning on the GPIIb/IIIa receptor, allowing these receptors to bind with vWF or fibrinogen.[6] There are around 60 000 of these receptors per platelet.[26] When any one or more of at least nine different platelet surface receptors are turned on during activation, intraplatelet signaling pathways cause existing GpIIb/IIIa receptors to change shape – curled to straight – and thus become capable of binding.[6]
Since fibrinogen is a rod-like protein with nodules on either end capable of binding GPIIb/IIIa, activated platelets with exposed GPIIb/IIIa can bind fibrinogen to aggregate. GPIIb/IIIa may also further anchor the platelets to subendothelial vWF for additional structural stabilisation.
Classically it was thought that this was the only mechanism involved in aggregation, but three new mechanisms have been identified which can initiate aggregation, depending on the velocity of blood flow (i.e. shear range).[27]
Wound repair
The blood clot is only a temporary solution to stop bleeding; tissue repair is needed. Small interruptions in the endothelium are handled by physiological mechanisms; large interruptions by the trauma surgeon.
[28] The fibrin is slowly dissolved by the fibrinolytic enzyme, plasmin, and the platelets are cleared by phagocytosis.[29]
Immune function
Platelets have central role in innate immunity, initiating and participating in multiple inflammatory processes, directly binding pathogens and even destroying them. This support clinical data which show that many with serious bacterial or viral infections have thrombocytopenia, thus reducing their contribution to inflammation. Also platelet-leukocyte aggregates (PLAs) found in circulation are typical in sepsis or inflammatory bowel disease, showing the connection between thrombocytes and immune cells sensu stricto.[30]
Immunothrombosis
As hemostasis is a basic function of thrombocytes in mammals, it also has its uses in possible infection confinement.[8] In case of injury, platelets, together with the coagulation cascade, form the first line of defense by forming a blood clot. Thus, hemostasis and host defense were intertwined in evolution. For example, in the Atlantic horseshoe crab (living fossil estimated to be over 400 million years old), the only blood cell type, the amebocyte, facilitates both the hemostatic function and the encapsulation and phagocytosis of pathogens by means of exocytosis of intracellular granules containing bactericidal defense molecules. Blood clotting supports the immune function by trapping the pathogenic bacteria within.[31]
Although thrombosis, blood coagulation in intact blood vessels, is usually viewed as a pathological immune response, leading to obturation of lumen of blood vessel and subsequent hypoxic tissue damage, in some cases, directed thrombosis, called immunothrombosis, can localy control the spread of the infection. The thrombosis is directed in concordance of platelets, neutrophils and monocytes. The process is initiated either by immune cells sensu stricto by activating their pattern recognition receptors (PRRs), or by platelet-bacterial binding. Platelets can bind to bacteria either directly through thrombocytic PRRs[30] and bacterial surface proteins, or via plasma proteins that bind both to platelets and bacteria.[32] Monocytes respond to bacterial pathogen-associated molecular patterns (PAMPs), or damage-associated molecular patterns (DAMPs) by activating the extrinsic pathway of coagulation. Neutrophils facilitate the blood coagulation by NETosis. In turn, the platelets facilitate neutrophils' NETosis. NETs bind tissue factor, binding the coagulation centres to the location of infection. They also activate the intrinsic coagulation pathway by providing its negatively charged surface to the factor XII. Other neutrophil secretions, such as proteolytic enzymes, which cleave coagulation inhibitors, also bolster the process.[8]
In case of inbalance throughout the regulation of immunothrombosis, this process can quickly become aberrant. Regulatory defects in immunothrombosis are suspected to be major factor in causing pathological thrombosis in many forms, such as disseminated intravascular coagulation (DIC) or deep vein thrombosis. DIC in sepsis is a prime example of both dysregulated coagulation process as well as undue systemic inflammatory response resulting in multitude of microthrombi of similar composition to that in physiological immunothrombosis - fibrin, platelets, neutrophils and NETs.[8]
Inflammation
Platelets are rapidly deployed to sites of injury or infection, and potentially modulate inflammatory processes by interacting with leukocytes and by secreting cytokines, chemokines and other inflammatory mediators.[33][34][35][36]
Platelets also secrete platelet-derived growth factor (PDGF).
Platelets modulate neutrophils by forming platelet-leukocyte aggregates (PLAs). These formation induce upregulated production of αmβ2 (Mac-1) integrin in neutrophils. Interaction with PLAs also induce degranulation and increased phagocytosis in neutrophils. Platelets are also the largest source of soluble CD40L which induces production of reactive oxygen species (ROS) and upregulate expression of adhesion molecules, such as E-selectin, ICAM-1 and VCAM-1, in neutrophils, activates macrophages and activates cytotoxic response in T and B lymphocytes.[30]
Recently, the dogma that mammalian platelets lacking nucleus are unable of autonomous locomotion was broken.[37] In fact, the platelets are active scavengers, scaling walls of blood vessels and reorganising the thrombus. They are able to recognize and adhere to many surfaces, including bacteria. They are even able to fully envelop them in their open canalicular system (OCP), leading to proposed name of the process being "covercytosis", rather than phagocytosis, as OCS is merely an invagination of outer plasma membrane. These platelet-bacteria bundles are then used as an interation platform for neutrophilsm which destroy the bacteria using the NETosis and phagocytosis.
Platelets also participate in chronic inflammatory diseases, such as synovitis or rheumatoid arthritis.[38] Platelets are activated by collagen receptor glycoprotein IV (GPVI). Proinflammatory platelet microvesicles trigger constant cytokine secretion from neighboring fibroblast-like synoviocytes, most prominently Il-6 and Il-8. Inflammatory damage to surrounding extracellular matrix continually reveals more collagen, maintaining the microvesicle production.
Adaptive immunity
Activated platelets are able to participate in adaptive immunity, interacting with antibodies. They are able to specifically bind IgG through FcγRIIA, receptor for constant fragment (Fc) of IgG. When activated and bound to IgG opsonised bacteria, the platelets subsequently release reactive oxygen species (ROS), antimicrobial peptides, defensins, kinocidins and proteases, killing the bacteria directly.[39] Platelets also secrete proinflammatory and procoagulant mediators such as inorganic polyphosphates or platelet factor 4 (PF4), connecting innate and adaptive immune responses.[39][40]
Signs and symptoms of disorders
Spontaneous and excessive bleeding can occur because of platelet disorders. This bleeding can be caused by deficient numbers of platelets, dysfunctional platelets, or very excessive numbers of platelets: over 1.0 million/microliter. (The excessive numbers create a relative von Willebrand factor deficiency due to sequestration.)[41][42]
One can get a clue as to whether bleeding is due to a platelet disorder or a coagulation factor disorder by the characteristics and location of the bleeding.[4]:815, Table 39-4 All of the following suggest platelet bleeding, not coagulation bleeding: the bleeding from a skin cut such as a razor nick is prompt and excessive, but can be controlled by pressure; spontaneous bleeding into the skin which causes a purplish stain named by its size: petechiae, purpura, ecchymoses; bleeding into mucous membranes causing bleeding gums, nose bleed, and gastrointestinal bleeding; menorrhagia; and intraretinal and intracranial bleeding.
Excessive numbers of platelets, and/or normal platelets responding to abnormal vessel walls, can result in venous thrombosis and arterial thrombosis. The symptoms depend on the site of thrombosis.
Tests of function
Bleeding time
Developed by Duke in 1910 and bearing his name, it measured the time for bleeding to stop from a standardized wound in the ear lobe which is blotted each 30 seconds. Normal was less than 3 minutes.[43] More modern techniques are now used. A normal bleeding time reflects sufficient platelet numbers and function plus normal microvascular.
Multiplate multiple electrode aggregometry
In the Multiplate analyzer, anticoagulated whole blood is mixed with saline and a platelet agonist in a single use cuvette with two pairs of electrodes. The increase in impedance between the electrodes as platelets aggregate onto them, is measured and visualized as a curve.[citation needed]
PFA-100
The PFA-100 (Platelet Function Assay-100) is a system for analysing platelet function in which citrated whole blood is aspirated through a disposable cartridge containing an aperture within a membrane coated with either collagen and epinephrine or collagen and ADP. These agonists induce platelet adhesion, activation and aggregation leading to rapid occlusion of the aperture and cessation of blood flow termed the closure time (CT). An elevated CT with EPI and collagen can indicate intrinsic defects such as von Willebrand disease, uremia, or circulating platelet inhibitors. The follow up test involving collagen and ADP is used to indicate if the abnormal CT with collagen and EPI was caused by the effects of acetyl sulfosalicylic acid (aspirin) or medications containing inhibitors.[44]
Disorders
Adapted from:[4]:vii
The three broad categories of platelet disorders are "not enough"; "dysfunctional"; and "too many".[4]:vii
Thrombocytopenia
Immune thrombocytopenias (ITP) – formerly known as immune thrombocytopenic purpura and idiopathic thrombocytopenic purpura
Splenomegaly- Gaucher's disease
- Familial thrombocytopenia[45]
- Chemotherapy
- Babesiosis
- Dengue
- Onyalai
- Thrombotic thrombocytopenic purpura
- HELLP syndrome
- Hemolytic-uremic syndrome
Drug-induced thrombocytopenic purpura (five known drugs – most problematic is heparin-induced thrombocytopenia (HIT)- Pregnancy associated
- Neonatal alloimmune associated
- Aplastic anemia
- Transfusion associated
- Pseudothrombocytopenia
- idiopathic thrombocytopenic purpura
Gilbert's Syndrome[46]
Altered platelet function
- Congenital
- Disorders of adhesion
- Bernard-Soulier syndrome
- Disorders of activation
- Disorders of granule amount or release
- Hermansky-Pudlak Syndrome
- Gray platelet syndrome
- ADP Receptor defect
- Decreased cyclooxygenase activity
- Storage pool defects, acquired or congenital
- Disorders of aggregation
- Glanzmann's thrombasthenia
- Wiskott–Aldrich syndrome
- Disorders of adhesion
- Acquired
- Disorders of adhesion
- Paroxysmal nocturnal hemoglobinuria
Asthma[47]
Samter's triad (aspirin exacerbated respiratory disease/AERD)[48]
Cancer[49]
Malaria[50]- Decreased cyclooxygenase activity
- Disorders of adhesion
Thrombocytosis and thrombocythemia
- Reactive
- Chronic infection
- Chronic inflammation
- Malignancy
- Hyposplenism (post-splenectomy)
- Iron deficiency
- Acute blood loss
Myeloproliferative neoplasms – platelets are both elevated and activated- Essential thrombocytosis
- Polycythemia vera
- Associated with other myeloid neoplasms
- Congenital
Drugs affecting
Anti-inflammatory drugs
Some drugs used to treat inflammation have the unwanted side effect of suppressing normal platelet function. These are the non-steroidal anti-inflammatory drugs (NSAIDS). Aspirin irreversibly disrupts platelet function by inhibiting cyclooxygenase-1 (COX1), and hence normal hemostasis. The resulting platelets are unable to produce new cyclooxygenase because they have no DNA. Normal platelet function will not return until the use of aspirin has ceased and enough of the affected platelets have been replaced by new ones, which can take over a week. Ibuprofen, another NSAID, does not have such a long duration effect, with platelet function usually returning within 24 hours,[51] and taking ibuprofen before aspirin prevents the irreversible effects of aspirin.[52]
Drugs that suppress platelet function
These drugs are used to prevent thrombus formation.
Oral agents
- aspirin
- clopidogrel
- cilostazol
- ticlopidine
- ticagrelor
- prasugrel
Drugs that stimulate platelet production
- thrombopoietin mimetics
- desmopressin
- factor VIIa
Intravenous agents
abciximab,- eptifibatide
- tirofiban
- Others oprelvekin, romiplostim, eltrombopag, argatroban
Therapy with platelets
Transfusion
Indications
Platelet transfusion is most frequently used to correct unusually low platelet counts, either to prevent spontaneous bleeding (typically at counts below 10×109/L) or in anticipation of medical procedures that will necessarily involve some bleeding. For example, in patients undergoing surgery, a level below 50×109/L is associated with abnormal surgical bleeding, and regional anaesthetic procedures such as epidurals are avoided for levels below 80×109/L.[53] Platelets may also be transfused when the platelet count is normal but the platelets are dysfunctional, such as when an individual is taking aspirin or clopidogrel.[54] Finally, platelets may be transfused as part of a massive transfusion protocol, in which the three major blood components (red blood cells, plasma, and platelets) are transfused to address severe hemorrhage. Platelet transfusion is contraindicated in thrombotic thrombocytopenic purpura (TTP), as it fuels the coagulopathy.
Collection

Platelet concentrate.
Platelets are either isolated from collected units of whole blood and pooled to make a therapeutic dose, or collected by platelet apheresis: blood is taken from the donor, passed through a device which removes the platelets, and the remainder is returned to the donor in a closed loop. The industry standard is for platelets to be tested for bacteria before transfusion to avoid septic reactions, which can be fatal. Recently the AABB Industry Standards for Blood Banks and Transfusion Services (5.1.5.1) has allowed for use of pathogen reduction technology as an alternative to bacterial screenings in platelets.[55]
Pooled whole-blood platelets, sometimes called “random” platelets, are separated by one of two methods.[56] In the US, a unit of whole blood is placed into a large centrifuge in what is referred to as a “soft spin.” At these settings, the platelets remain suspended in the plasma. The platelet-rich plasma (PRP) is removed from the red cells, then centrifuged at a faster setting to harvest the platelets from the plasma. In other regions of the world, the unit of whole blood is centrifuged using settings that cause the platelets to become suspended in the “buffy coat” layer, which includes the platelets and the white blood cells. The “buffy coat” is isolated in a sterile bag, suspended in a small amount of red blood cells and plasma, then centrifuged again to separate the platelets and plasma from the red and white blood cells. Regardless of the initial method of preparation, multiple donations may be combined into one container using a sterile connection device to manufacture a single product with the desired therapeutic dose.
Apheresis platelets are collected using a mechanical device that draws blood from the donor and centrifuges the collected blood to separate out the platelets and other components to be collected. The remaining blood is returned to the donor. The advantage to this method is that a single donation provides at least one therapeutic dose, as opposed to the multiple donations for whole-blood platelets. This means that a recipient is not exposed to as many different donors and has less risk of transfusion-transmitted disease and other complications. Sometimes a person such as a cancer patient who requires routine transfusions of platelets will receive repeated donations from a specific donor to further minimize the risk. Pathogen reduction of platelets using for example, riboflavin and UV light treatments can also be carried out to reduce the infectious load of pathogens contained in donated blood products, thereby reducing the risk of transmission of transfusion transmitted diseases.[57][58] Another photochemical treatment process utilizing amotosalen and UVA light has been developed for the inactivation of viruses, bacteria, parasites, and leukocytes that can contaminate blood components intended for transfusion.[59] In addition, apheresis platelets tend to contain fewer contaminating red blood cells because the collection method is more efficient than “soft spin” centrifugation at isolating the desired blood component.
Storage
Platelets collected by either method have a very short shelf life, typically five days. This results in frequent problems with short supply, as testing the donations often requires up to a full day. Since there are no effective preservative solutions for platelets, they lose potency quickly and are best when fresh.
Platelets are stored under constant agitation at 20–24 °C (68–75.2 °F). Units can not be refrigerated as this causes platelets to change shape and lose function. Storage at room temperature provides an environment where any bacteria that are introduced to the blood component during the collection process may proliferate and subsequently cause bacteremia in the patient. Regulations are in place in the United States that require products to be tested for the presence of bacterial contamination before transfusion.[60]
Delivery to recipients
Platelets do not need to belong to the same A-B-O blood group as the recipient or be cross-matched to ensure immune compatibility between donor and recipient unless they contain a significant amount of red blood cells (RBCs). The presence of RBCs imparts a reddish-orange color to the product, and is usually associated with whole-blood platelets. An effort is sometimes made to issue type specific platelets, but this is not critical as it is with RBCs.
Prior to issuing platelets to the recipient, they may be irradiated to prevent transfusion-associated graft versus host disease or they may be washed to remove the plasma if indicated.
The change in the recipient's platelet count after transfusion is termed the "increment" and is calculated by subtracting the pre-transfusion platelet count from the post-transfusion platelet count. Many factors affect the increment including the recipient's body size, the number of platelets transfused, and clinical features that may cause premature destruction of the transfused platelets. When recipients fail to demonstrate an adequate post-transfusion increment, this is termed platelet transfusion refractoriness.
Platelets, either apheresis-derived or random-donor, can be processed through a volume reduction process. In this process, the platelets are spun in a centrifuge and the excess plasma is removed, leaving 10 to 100 mL of platelet concentrate. Such volume-reduced platelets are normally transfused only to neonatal and pediatric patients, when a large volume of plasma could overload the child's small circulatory system. The lower volume of plasma also reduces the chances of an adverse transfusion reaction to plasma proteins.[61] Volume reduced platelets have a shelf life of only four hours.[62]
Wound therapy
Platelets release platelet-derived growth factor (PDGF), a potent chemotactic agent; and TGF beta, which stimulates the deposition of extracellular matrix; fibroblast growth factor, insulin-like growth factor 1, platelet-derived epidermal growth factor, and vascular endothelial growth factor. Local application of these factors in increased concentrations through Platelet-rich plasma (PRP) is used as an adjunct in wound healing.[63]
Other animals
Instead of having platelets, non-mammalian vertebrates have nucleated thrombocytes, which resemble B lymphocytes in morphology. They aggregate in response to thrombin, but not to ADP, serotonin, nor adrenaline, as platelets do.[64][65]
History
George Gulliver in 1841 drew pictures of platelets[66] using the twin lens (compound) microscope invented in 1830 by Joseph Jackson Lister.[67] This microscope improved resolution sufficiently to make it possible to see platelets for the first time.
William Addison in 1842 drew pictures of a platelet-fibrin clot.[68]
Lionel Beale in 1864 was the first to publish a drawing showing platelets.[69]
Max Schultze in 1865 described what he called "spherules", which he noted were much smaller than red blood cells, occasionally clumped, and were sometimes found in collections of fibrin material.[70]
Giulio Bizzozero in 1882 studied the blood of amphibians microscopically in vivo. He named Schultz's spherules (It.) piastrine: little plates.[71][72]
William Osler observed platelets and, in published lectures in 1886, called them a third corpuscle and a blood plaque; and described them as "a colorless protoplasmic disc".[73]
James Wright examined blood smears using the stain named for him, and used the term plates in his 1906 publication[74] but changed to platelets in his 1910 publication[75] which has become the universally accepted term.
The term thrombocyte (clot cell) came into use in the early 1900s and is sometimes used as a synonym for platelet; but not generally in the scientific literature, except as a root word for other terms related to platelets (e.g. thrombocytopenia meaning low platelets).[4]:v3 The term thrombocytes is proper for mononuclear cells found in the blood of non-mammalian vertebrates: they are the functional equivalent of platelets, but circulate as intact cells rather than cytoplasmic fragments of bone marrow megakaryocytes.[4]:3
In some contexts, the word thrombus is used interchangeably with the word clot, regardless of its composition (white, red, or mixed). In other contexts it is used to contrast a normal from an abnormal clot: thrombus arises from physiologic hemostasis, thrombosis arises from a pathologic and excessive quantity of clot.[76] In a third context it is used to contrast the result from the process: thrombus is the result, thrombosis is the process.
References
^ Laki K (December 1972). "Our ancient heritage in blood clotting and some of its consequences". Annals of the New York Academy of Sciences. 202 (1): 297–307. Bibcode:1972NYASA.202..297L. doi:10.1111/j.1749-6632.1972.tb16342.x. PMID 4508929..mw-parser-output cite.citationfont-style:inherit.mw-parser-output qquotes:"""""""'""'".mw-parser-output code.cs1-codecolor:inherit;background:inherit;border:inherit;padding:inherit.mw-parser-output .cs1-lock-free abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .cs1-lock-limited a,.mw-parser-output .cs1-lock-registration abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .cs1-lock-subscription abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registrationcolor:#555.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration spanborder-bottom:1px dotted;cursor:help.mw-parser-output .cs1-hidden-errordisplay:none;font-size:100%.mw-parser-output .cs1-visible-errorfont-size:100%.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-formatfont-size:95%.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-leftpadding-left:0.2em.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-rightpadding-right:0.2em
^ Machlus KR, Thon JN, Italiano JE (April 2014). "Interpreting the developmental dance of the megakaryocyte: a review of the cellular and molecular processes mediating platelet formation". British Journal of Haematology. 165 (2): 227–36. doi:10.1111/bjh.12758. PMID 24499183.
^ Jain NC (June 1975). "A scanning electron microscopic study of platelets of certain animal species". Thrombosis et Diathesis Haemorrhagica. 33 (3): 501–7. PMID 1154309.
^ abcdefg Michelson, Alan D. (2013). Platelets (3rd ed.). Academic. ISBN 9780123878373.
^ Paulus JM (September 1975). "Platelet size in man". Blood. 46 (3): 321–36. PMID 1097000.
^ abcde Yip J, Shen Y, Berndt MC, Andrews RK (February 2005). "Primary platelet adhesion receptors". IUBMB Life. 57 (2): 103–8. doi:10.1080/15216540500078962. PMID 16036569.
^ Berridge, Michael J. (1 October 2014). "Module 11: Cell Stress, Inflammatory Responses and Cell Death". Cell Signalling Biology. 6: csb0001011. doi:10.1042/csb0001011.
^ abcd Gaertner F, Massberg S (December 2016). "Blood coagulation in immunothrombosis-At the frontline of intravascular immunity". Seminars in Immunology. 28 (6): 561–569. doi:10.1016/j.smim.2016.10.010. PMID 27866916.
^ Hampton T (April 2018). "Platelets' Role in Adaptive Immunity May Contribute to Sepsis and Shock". JAMA. 319 (13): 1311–1312. doi:10.1001/jama.2017.12859. PMID 29614158.
^ Girling JH (July 1962). "An automatic platelet counting technique". The Journal of Medical Laboratory Technology. 19: 168–73. PMID 13898919.
^ Ross DW, Ayscue LH, Watson J, Bentley SA (September 1988). "Stability of hematologic parameters in healthy subjects. Intraindividual versus interindividual variation". American Journal of Clinical Pathology. 90 (3): 262–7. doi:10.1093/ajcp/90.3.262. PMID 3414599.
^ Ruocco L, Del Corso L, Romanelli AM, Deri D, Pentimone F (April 2001). "New hematological indices in the healthy elderly". Minerva Medica. 92 (2): 69–73. PMID 11323567.
^ Lozano M, Narváez J, Faúndez A, Mazzara R, Cid J, Jou JM, Marín JL, Ordinas A (June 1998). "[Platelet count and mean platelet volume in the Spanish population]". Medicina Clinica. 110 (20): 774–7. PMID 9666418.
^ Harker LA, Roskos LK, Marzec UM, Carter RA, Cherry JK, Sundell B, Cheung EN, Terry D, Sheridan W (April 2000). "Effects of megakaryocyte growth and development factor on platelet production, platelet life span, and platelet function in healthy human volunteers". Blood. 95 (8): 2514–22. PMID 10753829.
^ Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, Huang DC, Kile BT (March 2007). "Programmed anuclear cell death delimits platelet life span". Cell. 128 (6): 1173–86. doi:10.1016/j.cell.2007.01.037. PMID 17382885.
^ Palmer RM, Ferrige AG, Moncada S (1987). "Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor". Nature. 327 (6122): 524–6. Bibcode:1987Natur.327..524P. doi:10.1038/327524a0. PMID 3495737.
^ Jones CI, Barrett NE, Moraes LA, Gibbins JM, Jackson DE (2012). Endogenous inhibitory mechanisms and the regulation of platelet function. Methods in Molecular Biology. 788. pp. 341–66. doi:10.1007/978-1-61779-307-3_23. ISBN 978-1-61779-306-6. PMID 22130718.
^ Marcus AJ, Broekman MJ, Drosopoulos JH, Olson KE, Islam N, Pinsky DJ, Levi R (April 2005). "Role of CD39 (NTPDase-1) in thromboregulation, cerebroprotection, and cardioprotection". Seminars in Thrombosis and Hemostasis. 31 (2): 234–46. doi:10.1055/s-2005-869528. PMID 15852226.
^ Dubois C, Panicot-Dubois L, Merrill-Skoloff G, Furie B, Furie BC (May 2006). "Glycoprotein VI-dependent and -independent pathways of thrombus formation in vivo". Blood. 107 (10): 3902–6. doi:10.1182/blood-2005-09-3687. PMC 1895285. PMID 16455953.
^ Matarrese P, Straface E, Palumbo G, Anselmi M, Gambardella L, Ascione B, Del Principe D, Malorni W (February 2009). "Mitochondria regulate platelet metamorphosis induced by opsonized zymosan A--activation and long-term commitment to cell death". The FEBS Journal. 276 (3): 845–56. doi:10.1111/j.1742-4658.2008.06829.x. PMID 19143843.
^ White JG (December 1987). "An overview of platelet structural physiology". Scanning Microsc. 1 (4): 1677–700. PMID 3324323.
^ Behnke O (1970). "The morphology of blood platelet membrane systems". Series Haematologica. 3 (4): 3–16. PMID 4107203.
^ ab Bouchard BA, Mann KG, Butenas S (August 2010). "No evidence for tissue factor on platelets". Blood. 116 (5): 854–5. doi:10.1182/blood-2010-05-285627. PMC 2918337. PMID 20688968.
^ Ahmad SS, Rawala-Sheikh R, Walsh PN (1992). "Components and assembly of the factor X activating complex". Seminars in Thrombosis and Hemostasis. 18 (3): 311–23. doi:10.1055/s-2007-1002570. PMID 1455249.
^ Tyagi T, Ahmad S, Gupta N, Sahu A, Ahmad Y, Nair V, Chatterjee T, Bajaj N, Sengupta S, Ganju L, Singh SB, Ashraf MZ (February 2014). "Altered expression of platelet proteins and calpain activity mediate hypoxia-induced prothrombotic phenotype". Blood. 123 (8): 1250–60. doi:10.1182/blood-2013-05-501924. PMID 24297866.
^ O'Halloran AM, Curtin R, O'Connor F, Dooley M, Fitzgerald A, O'Brien JK, Fitzgerald DJ, Shields DC (February 2006). "The impact of genetic variation in the region of the GPIIIa gene, on Pl expression bias and GPIIb/IIIa receptor density in platelets". British Journal of Haematology. 132 (4): 494–502. doi:10.1111/j.1365-2141.2005.05897.x. PMID 16412022.
^ Coller BS, Cheresh DA, Asch E, Seligsohn U (January 1991). "Platelet vitronectin receptor expression differentiates Iraqi-Jewish from Arab patients with Glanzmann thrombasthenia in Israel". Blood. 77 (1): 75–83. PMID 1702031.
^ Nguyen, D.T., Orgill D.P., Murphy G.F. (2009). Chapter 4: The Pathophysiologic Basis for Wound Healing and Cutaneous Regeneration. Biomaterials For Treating Skin Loss. Woodhead Publishing (UK/Europe) & CRC Press (US), Cambridge/Boca Raton, pp. 25–57. (
ISBN 978-1-4200-9989-8
ISBN 978-1-84569-363-3)
^ Movat HZ, Weiser WJ, Glynn MF, Mustard JF (December 1965). "Platelet phagocytosis and aggregation". The Journal of Cell Biology. 27 (3): 531–43. doi:10.1083/jcb.27.3.531. PMC 2106759. PMID 4957257.
^ abc Jenne CN, Urrutia R, Kubes P (June 2013). "Platelets: bridging hemostasis, inflammation, and immunity". International Journal of Laboratory Hematology. 35 (3): 254–61. doi:10.1111/ijlh.12084. PMID 23590652.
^ Levin J (2007), "The Evolution of Mammalian Platelets", Platelets, Elsevier, pp. 3–22, doi:10.1016/b978-012369367-9/50763-1, ISBN 9780123693679
^ Cox D, Kerrigan SW, Watson SP (June 2011). "Platelets and the innate immune system: mechanisms of bacterial-induced platelet activation". Journal of Thrombosis and Haemostasis. 9 (6): 1097–107. doi:10.1111/j.1538-7836.2011.04264.x. PMID 21435167.
^ Weyrich AS, Zimmerman GA (September 2004). "Platelets: signaling cells in the immune continuum". Trends in Immunology. 25 (9): 489–95. doi:10.1016/j.it.2004.07.003. PMID 15324742.
^ Wagner DD, Burger PC (December 2003). "Platelets in inflammation and thrombosis". Arteriosclerosis, Thrombosis, and Vascular Biology. 23 (12): 2131–7. doi:10.1161/01.ATV.0000095974.95122.EC. PMID 14500287.
^ Diacovo TG, Puri KD, Warnock RA, Springer TA, von Andrian UH (July 1996). "Platelet-mediated lymphocyte delivery to high endothelial venules". Science. 273 (5272): 252–5. Bibcode:1996Sci...273..252D. doi:10.1126/science.273.5272.252. PMID 8662511.
^ Iannacone M, Sitia G, Isogawa M, Marchese P, Castro MG, Lowenstein PR, Chisari FV, Ruggeri ZM, Guidotti LG (November 2005). "Platelets mediate cytotoxic T lymphocyte-induced liver damage". Nature Medicine. 11 (11): 1167–9. doi:10.1038/nm1317. PMC 2908083. PMID 16258538.
^ Gaertner F, Ahmad Z, Rosenberger G, Fan S, Nicolai L, Busch B, Yavuz G, Luckner M, Ishikawa-Ankerhold H, Hennel R, Benechet A, Lorenz M, Chandraratne S, Schubert I, Helmer S, Striednig B, Stark K, Janko M, Böttcher RT, Verschoor A, Leon C, Gachet C, Gudermann T, Mederos Y, Schnitzler M, Pincus Z, Iannacone M, Haas R, Wanner G, Lauber K, Sixt M, Massberg S (November 2017). "Migrating Platelets Are Mechano-scavengers that Collect and Bundle Bacteria". Cell. 171 (6): 1368–1382.e23. doi:10.1016/j.cell.2017.11.001. PMID 29195076.
^ Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, Massarotti EM, Remold-O'Donnell E, Farndale RW, Ware J, Lee DM (January 2010). "Platelets amplify inflammation in arthritis via collagen-dependent microparticle production". Science. 327 (5965): 580–3. Bibcode:2010Sci...327..580B. doi:10.1126/science.1181928. PMC 2927861. PMID 20110505.
^ ab Palankar R, Kohler TP, Krauel K, Wesche J, Hammerschmidt S, Greinacher A (June 2018). "Platelets kill bacteria by bridging innate and adaptive immunity via platelet factor 4 and FcγRIIA". Journal of Thrombosis and Haemostasis. 16 (6): 1187–1197. doi:10.1111/jth.13955. PMID 29350833.
^ McMorran BJ, Wieczorski L, Drysdale KE, Chan JA, Huang HM, Smith C, Mitiku C, Beeson JG, Burgio G, Foote SJ (December 2012). "Platelet factor 4 and Duffy antigen required for platelet killing of Plasmodium falciparum". Science. 338 (6112): 1348–51. Bibcode:2012Sci...338.1348M. doi:10.1126/science.1228892. PMID 23224555.
^ Murakawa M, Okamura T, Tsutsumi K, Tanoguchi S, Kamura T, Shibuya T, Harada M, Niho Y (1992). "Acquired von Willebrand's disease in association with essential thrombocythemia: regression following treatment". Acta Haematologica. 87 (1–2): 83–7. doi:10.1159/000204725. PMID 1585777.
^ van Genderen PJ, Leenknegt H, Michiels JJ, Budde U (September 1996). "Acquired von Willebrand disease in myeloproliferative disorders". Leukemia & Lymphoma. 22 Suppl 1: 79–82. doi:10.3109/10428199609074364. PMID 8951776.
^ Duke WW (1910). "The relation of blood platelets to hemorrhagic disease". JAMA. 55 (14): 1185–92. doi:10.1001/jama.1910.04330140029009.
^ "Platelet Function Assay FAQ" (PDF). Department of Pathology. Virginia Commonwealth University. Retrieved 2017-03-27.
^ Geddis AE (February 2013). "Inherited thrombocytopenias: an approach to diagnosis and management". International Journal of Laboratory Hematology. 35 (1): 14–25. doi:10.1111/j.1751-553x.2012.01454.x. PMID 22846067.
^ Cure MC, Cure E, Kirbas A, Cicek AC, Yuce S (July 2013). "The effects of Gilbert's syndrome on the mean platelet volume and other hematological parameters". Blood Coagulation & Fibrinolysis. 24 (5): 484–8. doi:10.1097/MBC.0b013e32835e4230. PMID 23348429.
^ Kornerup KN, Page CP (August 2007). "The role of platelets in the pathophysiology of asthma". Platelets. 18 (5): 319–28. doi:10.1080/09537100701230436. PMID 17654302.
^ Laidlaw TM, Kidder MS, Bhattacharyya N, Xing W, Shen S, Milne GL, Castells MC, Chhay H, Boyce JA (April 2012). "Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes". Blood. 119 (16): 3790–8. doi:10.1182/blood-2011-10-384826. PMC 3335383. PMID 22262771.
^ Erpenbeck L, Schön MP (April 2010). "Deadly allies: the fatal interplay between platelets and metastasizing cancer cells". Blood. 115 (17): 3427–36. doi:10.1182/blood-2009-10-247296. PMC 2867258. PMID 20194899.
^ Pleass RJ (July 2009). "Platelet power: sticky problems for sticky parasites?". Trends in Parasitology. 25 (7): 296–9. doi:10.1016/j.pt.2009.04.002. PMC 3116138. PMID 19539528.
^ "Summaries for patients. Platelet function after taking Ibuprofen for 1 week". Annals of Internal Medicine. 142 (7): I-54. April 2005. doi:10.7326/0003-4819-142-7-200504050-00004. PMID 15809457.
^ Rao GH, Johnson GG, Reddy KR, White JG (1983). "Ibuprofen protects platelet cyclooxygenase from irreversible inhibition by aspirin". Arteriosclerosis. 3 (4): 383–8. doi:10.1161/01.ATV.3.4.383. PMID 6411052.
^ van Veen JJ, Nokes TJ, Makris M (January 2010). "The risk of spinal haematoma following neuraxial anaesthesia or lumbar puncture in thrombocytopenic individuals". British Journal of Haematology. 148 (1): 15–25. doi:10.1111/j.1365-2141.2009.07899.x. PMID 19775301.
^ Roback J, Grossman B, Harris T, Hillyer C, eds. (2011). Technical Manual (17th ed.). Bethesda MD: AABB. p. 580. ISBN 978-1-56395-315-6.
^ American Association of Blood Banks (2003). "5.1.5.1". Standards for Blood Banks and Transfusion Services (22nd ed.). Bethesda MD: AABB.
^ Högman CF (January 1992). "New trends in the preparation and storage of platelets". Transfusion. 32 (1): 3–6. doi:10.1046/j.1537-2995.1992.32192116428.x. PMID 1731433.
^ Ruane PH, Edrich R, Gampp D, Keil SD, Leonard RL, Goodrich RP (June 2004). "Photochemical inactivation of selected viruses and bacteria in platelet concentrates using riboflavin and light". Transfusion. 44 (6): 877–85. doi:10.1111/j.1537-2995.2004.03355.x. PMID 15157255.
^ Perez-Pujol S, Tonda R, Lozano M, Fuste B, Lopez-Vilchez I, Galan AM, Li J, Goodrich R, Escolar G (June 2005). "Effects of a new pathogen-reduction technology (Mirasol PRT) on functional aspects of platelet concentrates". Transfusion. 45 (6): 911–9. doi:10.1111/j.1537-2995.2005.04350.x. PMID 15934989.
^ Prowse CV (April 2013). "Component pathogen inactivation: a critical review". Vox Sanguinis. 104 (3): 183–99. doi:10.1111/j.1423-0410.2012.01662.x. PMID 23134556.
^ AABB (2009). Standards for Blood Banks and Transfusion Services (26th ed.). Bethesda MD: AABB.
^ Schoenfeld H, Spies C, Jakob C (March 2006). "Volume-reduced platelet concentrates". Current Hematology Reports. 5 (1): 82–8. PMID 16537051.
^ CBBS: Washed and volume-reduced Plateletpheresis units. Cbbsweb.org (2001-10-25). Retrieved on 2011-11-14.
^ Gawaz M, Vogel S (October 2013). "Platelets in tissue repair: control of apoptosis and interactions with regenerative cells". Blood. 122 (15): 2550–4. doi:10.1182/blood-2013-05-468694. PMID 23963043.
^ Schmaier AA, Stalker TJ, Runge JJ, Lee D, Nagaswami C, Mericko P, Chen M, Cliché S, Gariépy C, Brass LF, Hammer DA, Weisel JW, Rosenthal K, Kahn ML (September 2011). "Occlusive thrombi arise in mammals but not birds in response to arterial injury: evolutionary insight into human cardiovascular disease". Blood. 118 (13): 3661–9. doi:10.1182/blood-2011-02-338244. PMC 3186337. PMID 21816834.
^ Belamarich FA, Shepro D, Kien M (November 1968). "ADP is not involved in thrombin-induced aggregation of thrombocytes of a non-mammalian vertebrate". Nature. 220 (5166): 509–10. Bibcode:1968Natur.220..509B. doi:10.1038/220509a0. PMID 5686175.
^ Lancet, 1882, ii. 916; Notes of Gulliver's Researches in Anatomy, Physiology, Pathology, and Botany, 1880; Carpenter's Physiology, ed. Power, 9th ed., see Index under 'Gulliver.'
^
Godlee, Sir Rickman (1917). Lord Lister. London: Macmillan & Co.
^ Robb-Smith AH (July 1967). "Why the platelets were discovered". British Journal of Haematology. 13 (4): 618–37. doi:10.1111/j.1365-2141.1967.tb00769.x. PMID 6029960.
^ Beale LS (1864). "On the Germinal Matter of the Blood, with Remarks upon the Formation of Fibrin". Transactions of the Microscopical Society & Journal. 12: 47–63. doi:10.1111/j.1365-2818.1864.tb01625.x.
^ Schultze M (1865). "Ein heizbarer Objecttisch und seine Verwendung bei Untersuchungen des Blutes". Arch Mikrosk Anat. 1 (1): 1–42. doi:10.1007/BF02961404.
^ Bizzozero, J. (1882). "Über einen neuen Forrnbestandteil des Blutes und dessen Rolle bei der Thrombose und Blutgerinnung". Arch Pathol Anat Phys Klin Med. 90 (2): 261–332. doi:10.1007/BF01931360.
^ Brewer DB (May 2006). "Max Schultze (1865), G. Bizzozero (1882) and the discovery of the platelet". British Journal of Haematology. 133 (3): 251–8. doi:10.1111/j.1365-2141.2006.06036.x. PMID 16643426.
^ Osler W (1886). "On certain problems in the physiology of the blood corpuscles". The Medical News. 48: 421–25.
^ Wright JH (1906). "The Origin and Nature of the Blood Plates". The Boston Medical and Surgical Journal. 154 (23): 643–45. doi:10.1056/NEJM190606071542301.
^ Wright JH (1910). "The histogenesis of blood platelets". Journal of Morphology. 21 (2): 263–78. doi:10.1002/jmor.1050210204. hdl:2027/hvd.32044107223588.
^ Furie B, Furie BC (August 2008). "Mechanisms of thrombus formation". The New England Journal of Medicine. 359 (9): 938–49. doi:10.1056/NEJMra0801082. PMID 18753650.
External links
| Wikimedia Commons has media related to platelets. |
Video summarizing platelet dynamics ( Page will play audio when loaded)
Page will play audio when loaded)
Authority control |
|
|---|


