Potential energy surface
A potential energy surface (PES) describes the energy of a system, especially a collection of atoms, in terms of certain parameters, normally the positions of the atoms. The surface might define the energy as a function of one or more coordinates; if there is only one coordinate, the surface is called a potential energy curve or energy profile. An example is the Morse/Long-range potential.
It is helpful to use the analogy of a landscape: for a system with two degrees of freedom (e.g. two bond lengths), the value of the energy (analogy: the height of the land) is a function of two bond lengths (analogy: the coordinates of the position on the ground).[1]
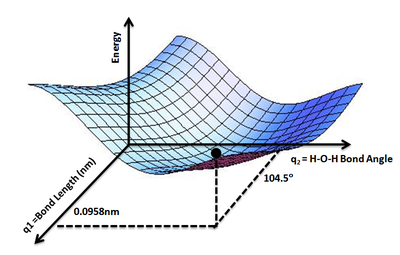
PES for water molecule: Shows the energy minimum corresponding to optimized molecular structure for water- O-H bond length of 0.0958nm and H-O-H bond angle of 104.5°
The PES concept finds application in fields such as chemistry and physics, especially in the theoretical sub-branches of these subjects. It can be used to theoretically explore properties of structures composed of atoms, for example, finding the minimum energy shape of a molecule or computing the rates of a chemical reaction.
Contents
1 Mathematical definition and computation
2 Application
3 Attractive and repulsive surfaces
4 History
5 See also
6 References
Mathematical definition and computation
The geometry of a set of atoms can be described by a vector, r, whose elements represent the atom positions. The vector r could be the set of the Cartesian coordinates of the atoms, or could also be a set of inter-atomic distances and angles.
Given r, the energy as a function of the positions, E(r), is the value of E(r) for all r of interest. Using the landscape analogy from the introduction, E gives the height on the "energy landscape" so that the concept of a potential energy surface arises.
To study a chemical reaction using the PES as a function of atomic positions, it is necessary to calculate the energy for every atomic arrangement of interest. Methods of calculating the energy of a particular atomic arrangement of atoms are well described in the computational chemistry article, and the emphasis here will be on finding approximations of E(r) to yield fine-grained energy-position information.
For very simple chemical systems or when simplifying approximations are made about inter-atomic interactions, it is sometimes possible to use an analytically derived expression for the energy as a function of the atomic positions. An example is the London-Eyring-Polanyi-Sato potential[2][3][4] for the system H + H2 as a function of the three H-H distances.
For more complicated systems, calculation of the energy of a particular arrangement of atoms is often too computationally expensive for large scale representations of the surface to be feasible. For these systems a possible approach is to calculate only a reduced set of points on the PES and then use a computationally cheaper interpolation method, for example Shepard interpolation, to fill in the gaps.[5]
Application
A PES is a conceptual tool for aiding the analysis of molecular geometry and chemical reaction dynamics. Once the necessary points are evaluated on a PES, the points can be classified according to the first and second derivatives of the energy with respect to position, which respectively are the gradient and the curvature. Stationary points (or points with a zero gradient) have physical meaning: energy minima correspond to physically stable chemical species and saddle points correspond to transition states, the highest energy point on the reaction coordinate (which is the lowest energy pathway connecting a chemical reactant to a chemical product).
Attractive and repulsive surfaces
Potential energy surfaces for chemical reactions can be classified as attractive or repulsive by comparing the extensions of the bond lengths in the activated complex relative to those of the reactants and products.[6][7] For a reaction of type A + B—C → A—B + C, the bond length extension for the newly formed A—B bond is defined as R*AB = RAB − R0AB, where RAB is the A—B bond length in the transition state and R0AB in the product molecule. Similarly for the bond which is broken in the reaction, R*BC = RBC − R0BC, where R0BC refers to the reactant molecule.[8]
For exothermic reactions, a PES is classified as attractive (or early-downhill) if R*AB > R*BC, so that the transition state is reached while the reactants are approaching each other. After the transition state, the A—B bond length continues to decrease, so that much of the liberated reaction energy is converted into vibrational energy of the A—B bond.[8][9] An example is the harpoon reaction K + Br2 → K—Br + Br, in which the initial long-range attraction of the reactants leads to an activated complex resembling K+•••Br−•••Br.[8] The vibrationally excited populations of product molecules can be detected by infrared chemiluminescence.[10][11]
In contrast the PES for the reaction H + Cl2 → HCl + Cl is repulsive (or late-downhill) because R*HCl < R*ClCl and the transition state is reached when the products are separating.[8][9] For this reaction in which the atom A (here H) is lighter than B and C, the reaction energy is released primarily as translational kinetic energy of the products.[8] For a reaction such as F + H2 → HF + H in which atom A is heavier than B and C, there is mixed energy release, both vibrational and translational, even though the PES is repulsive.[8]
For endothermic reactions, the type of surface determines the type of energy which is most effective in bringing about reaction. Translational energy of the reactants is most effective at inducing reactions with an attractive surface, while vibrational excitation is more effective for reactions with a repulsive surface.[8] As an example of the latter case, the reaction F + HCl(v=1)[12] → Cl + HF is about five times faster than F + HCl(v=0) → Cl + HF for the same total energy of HCl.[13]
History
The concept of a potential energy surface for chemical reactions was first suggested by the French physicist René Marcelin in 1913.[14] The first semi-empirical calculation of a potential energy surface was proposed for the H + H2 reaction by Henry Eyring and Michael Polanyi in 1931. Eyring used potential energy surfaces to calculate reaction rate constants in the transition state theory in 1935.
See also
- Computational chemistry
Energy minimization (or geometry optimization)- Energy profile (chemistry)
- Reaction coordinate
References
^ Potential-energy (reaction) surface in Compendium of Chemical Terminology, 2nd ed. (the "Gold Book"). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997)
^ Sato, S. (1955). "A New Method of Drawing the Potential Energy Surface". Bulletin of the Chemical Society of Japan. 28 (7): 450. doi:10.1246/bcsj.28.450..mw-parser-output cite.citationfont-style:inherit.mw-parser-output .citation qquotes:"""""""'""'".mw-parser-output .citation .cs1-lock-free abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .citation .cs1-lock-limited a,.mw-parser-output .citation .cs1-lock-registration abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .citation .cs1-lock-subscription abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registrationcolor:#555.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration spanborder-bottom:1px dotted;cursor:help.mw-parser-output .cs1-ws-icon abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/4/4c/Wikisource-logo.svg/12px-Wikisource-logo.svg.png")no-repeat;background-position:right .1em center.mw-parser-output code.cs1-codecolor:inherit;background:inherit;border:inherit;padding:inherit.mw-parser-output .cs1-hidden-errordisplay:none;font-size:100%.mw-parser-output .cs1-visible-errorfont-size:100%.mw-parser-output .cs1-maintdisplay:none;color:#33aa33;margin-left:0.3em.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-formatfont-size:95%.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-leftpadding-left:0.2em.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-rightpadding-right:0.2em
"On a New Method of Drawing the Potential Energy Surface". The Journal of Chemical Physics. 23 (3): 592. 1955. Bibcode:1955JChPh..23..592S. doi:10.1063/1.1742043.
^ Keith J. Laidler, Chemical Kinetics (3rd ed., Harper & Row 1987) p.68-70
ISBN 0-06-043862-2
^ Steinfeld J.I., Francisco J.S. and Hase W.L. Chemical Kinetics and Dynamics (2nd ed., Prentice-Hall 1998) p.201-2
ISBN 0-13-737123-3
^ Moving least-squares enhanced Shepard interpolation for the fast marching and string methods, Burger SK1, Liu Y, Sarkar U, Ayers PW, J Chem Phys. 2009 130(2) 024103. doi: 10.1063/1.2996579.
^ Attractive potential-energy surface in Compendium of Chemical Terminology, 2nd ed. (the "Gold Book"). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997)
^ Repulsive potential-energy surface in Compendium of Chemical Terminology, 2nd ed. (the "Gold Book"). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997)
^ abcdefg Keith J. Laidler, Chemical Kinetics (3rd ed., Harper & Row 1987) p.461-8
ISBN 0-06-043862-2
^ ab Steinfeld J.I., Francisco J.S. and Hase W.L. Chemical Kinetics and Dynamics (2nd ed., Prentice-Hall 1998) p.272-4
ISBN 0-13-737123-3
^ Steinfeld J.I., Francisco J.S. and Hase W.L. Chemical Kinetics and Dynamics (2nd ed., Prentice-Hall 1998) p.263
ISBN 0-13-737123-3
^ Atkins P. and de Paula J. Physical Chemistry (8th ed., W.H.Freeman 2006) p.886
ISBN 0-7167-8759-8
^ Here v is the vibratonal quantum number.
^ Atkins P. and de Paula J. Physical Chemistry (8th ed., W.H.Freeman 2006) p.889-890
ISBN 0-7167-8759-8
^ Computational Chemistry: Introduction to the Theory and Applications of Molecular and Quantum Mechanics Errol G. Lewars, 2nd ed. (Springer 2011) p.21
ISBN 978-9048138616


